

The Osteopetrosis Support Trust - an Overview of Osteopetrosis
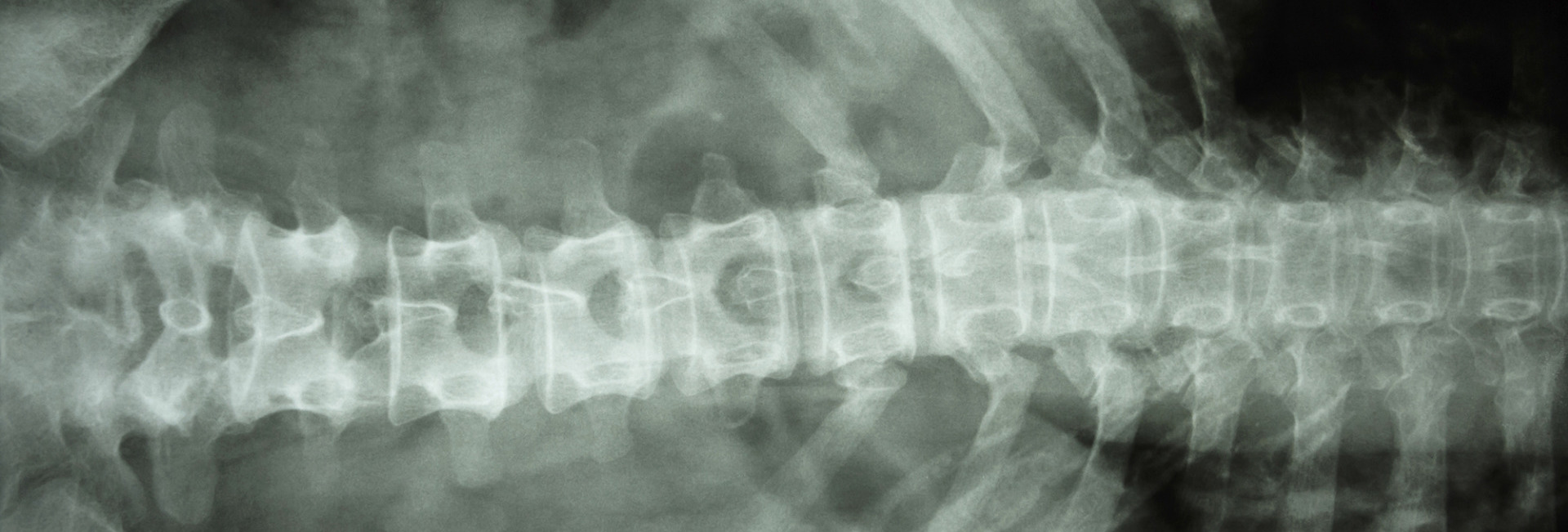
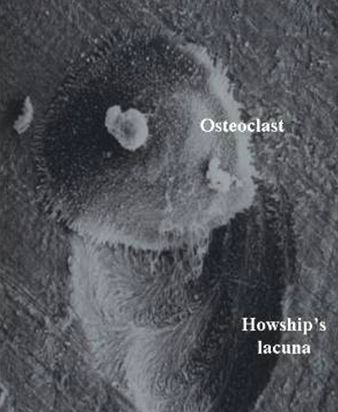
Osteopetrosis is general name for a group of rare genetic diseases all which are characterised by increased bone density. These result from problems in formation or function of osteoclasts, cells which dissolve bone so that it can be reshaped during growth or repaired after injury. The dense bones are prone to fracturing easily. Also, nerves which pass through channels in bone (such as those which pass through the skull to the eye or ear) can be damaged, since these channels do not enlarge as the nerves grow.
Medical text provided by Dr Colin Steward, now retired Consultant in BMT for Metabolic & Genetic Diseases.
What Are the Symptoms of Osteopetrosis?
Infantile ('Malignant') Osteopetrosis (MIOP) is the most severe form of the disease. Affected children tend to be short but with relatively large heads. They often sustain birth fractures and may be jittery or have convulsions during the first month of life due to a low blood calcium (Hypocalcaemia). Subsequently, they develop persistent snuffles (due to narrow nasal passages) and are prone to suffer increased ear, nose and throat, and respiratory infections. Bone marrow cavities are reduced: blood then accumulates in the liver and spleen, causing swelling of these organs and a low blood count may result. Many children become blind by six months of age, deafness and Hydrocephalus may develop later. Tooth eruption may be delayed and tooth quality tends to be poor.
Adult ('Benign') Osteopetrosis is most commonly diagnosed in late adolescence or early adulthood when bones are X-rayed due to a fracture, although it can sometimes be diagnosed in younger children especially where it is an incidental finding on an X-ray. Some people may go on to develop bone pain, dental problems and deafness or facial nerve paralysis. There is currently no specific treatment.
Inheritance Patterns & Prenatal Diagnosis
Inheritance Patterns
There has been rapid increase in the understanding of the genetics of Osteopetrosis since 2000. It is now known that mutation in any one of nine genes (carbonic anhydrase [CAII], proton pump [TCIRG1], chloride channel [ClCN7], OSTM1, RANK [TNFRSF11A], RANK Ligand [TNFSF11], cathepsin K [CTSK], PLEKHM1 and NF-kB Essential Modulator [NEMO]) is responsible for three quarters of cases, with no causative gene having so far been identified in the remaining quarter. Autosomal Recessive Disease may result from mutation of any of these genes. 50 per cent of babies/infants with Osteopetrosis will have mutations of TCIRG1 and 15 per cent will have mutations of ClCN7.
Adult Osteopetrosis is usually due to dominant mutations of ClCN7, and is therefore termed Autosomal Dominant Osteopetrosis (ADO).
Prenatal Diagnosis
Accurate antenatal diagnosis is possible in families with known gene mutations by either Chorionic Villus Sampling or Amniocentesis from ten weeks of pregnancy onwards. For other families prenatal diagnosis is less reliable and depends on detection of changes in the fetus by X-ray or ultrasound in the last third of pregnancy, both being tests which may still fail to detect accurately affected fetuses.

How Is It Treated?
Without treatment many affected children die before adolescence. However, at least half of children affected by the disease can be helped by Haemopoietic Stem Cell Transplantation (HSCT) using bone marrow, cord blood or peripheral blood stem cells from family or unrelated donors. This tends to be most widely used in patients with TCIRG1 mutations. Severe brain problems can develop some time after otherwise successful transplants in children with ClCN7 mutations and always develop in those with OSTM1 mutations. Transplantation therefore has an unclear role in ClCN7 disease and must not be performed in OSTM1 disease. It also does not work for children whose disease is caused by TNFSF11 mutations and has not been reported in ADO disease. Expert assessment and genetic analysis is therefore important if inappropriate transplantation is to be avoided. Injections of Gamma Interferon may be effective in alleviating some symptoms of the disease but are not widely used.